Your 2026 Guide to ISO 13485 Medical Devices
Your team has a promising prototype on the bench. The CAD is clean, the fit looks right, and the first CNC or printed parts are close enough to keep momentum. Then the important questions start. Which records do you need to keep? Can you use a general machine shop for verification builds? Does your prototype supplier need ISO 13485 certification, or just good process control? What changes once the device moves from concept work into formal validation?
That's where most medical device programs either get disciplined or get expensive. The issue usually isn't that engineers can't design the part. It's that design decisions, prototype choices, inspection plans, and supplier controls all start carrying regulatory weight long before production release.
In practice, ISO 13485 for medical devices matters most when it reaches the shop floor. It affects how you define tolerances, how you document surface finish requirements, how you approve suppliers, and whether the parts used in verification and validation can stand up to review later. If you treat it as paperwork added after the build, you create rework. If you use it as a framework during development, you reduce avoidable resets.
Table of Contents
- Navigating Compliance in Medical Device Development
- What Is ISO 13485 and Why Does It Matter
Who it applies to in real programs
Why teams care before certification
Understanding the Key Requirements of ISO 13485- Risk control starts with design decisions
- The Medical Device File has to match the physical part
- Process validation and traceability show up earlier than most teams expect
ISO 13485 vs ISO 9001 and Regulatory Frameworks- Why ISO 9001 does not answer the medical device question
- How ISO 13485 relates to FDA and EU requirements
- What this means during prototyping and supplier selection
An Implementation Checklist for ISO 13485- Phase one sets the pace
Supplier and Prototyping Implications- What to ask a prototype supplier
The Audit and Certification Process Explained- What the initial certification cycle looks like
Navigating Compliance in Medical Device Development
Medical device development gets complicated the moment a part stops being “just a prototype” and starts becoming evidence. That shift happens earlier than many teams expect. A printed enclosure used for ergonomic feedback is one thing. A machined component used in design verification is something else entirely.
The practical challenge is that engineering, supply chain, manufacturing, and quality often move at different speeds. Engineering wants iteration. Purchasing wants lead time certainty. Quality wants documented control. Regulatory wants objective evidence that the finished device will be safe and perform as intended. None of those priorities are wrong, but they collide fast if the team doesn't work from one quality framework.
ISO 13485 gives that framework. It's less useful to think of it as a certification target and more useful to treat it as the operating model for how a medical device organization designs, documents, builds, inspects, and releases product.
Practical rule: If a build may later support verification, validation, or design transfer, control it from the start as if someone will ask for the records later. They usually will.
Teams that struggle with compliance often make the same mistake. They separate technical development from quality system development. In practice, those are the same project. The tolerance stack-up, the inspection method, the supplier approval, and the revision history all become part of one traceable story.
That story is what gets a device through audits, customer reviews, and market entry without unnecessary backtracking.
What Is ISO 13485 and Why Does It Matter
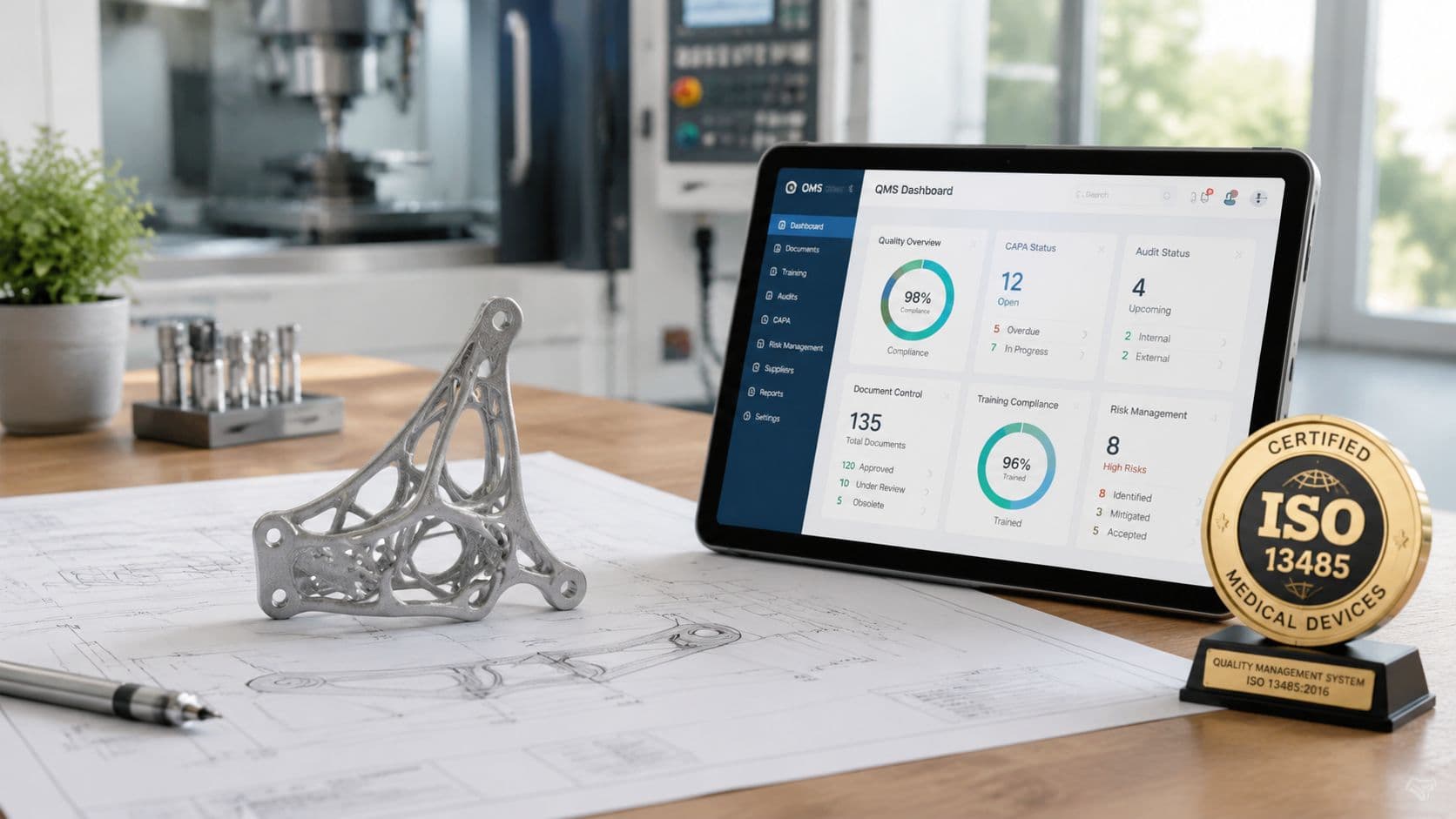
A team sends out a prototype housing for CNC machining, gets parts back in a week, runs bench testing, and then realizes nobody can clearly show which revision was built, which inspection results apply, or whether the supplier was approved for that kind of work. That is the point where ISO 13485 stops looking like a certification topic and starts looking like basic project control.
ISO 13485 is the quality management system standard written for medical device organizations. The current version, ISO 13485:2016, defines how a company controls design, purchasing, production, inspection, release, and post-delivery activities so the physical product matches its documented requirements.
For engineering teams, that matters early. The standard affects whether a 3D print is only a form model or can support verification work. It affects whether a low-volume machined lot needs first article data, material certs, and traceable revision control. It affects whether a supplier is just making parts or contributing to a controlled process that may later support submission, audit, or transfer to production.
Who it applies to in real programs
ISO 13485 reaches beyond the legal manufacturer. It applies across the companies and functions involved in design, production, installation, servicing, and related support activities. In practice, the OEM carries the final responsibility, but suppliers still matter because their process controls and records become part of the evidence chain.
That supplier point is often misunderstood.
A machine shop does not automatically need its own ISO 13485 certificate to cut prototype parts for a medical device developer. In many cases, the OEM can use a non-certified supplier if the supplier is properly qualified, the scope is appropriate, and the incoming product is controlled to the level the project requires. Once a supplier performs work that affects device conformity in a more direct or repeatable way, the qualification bar usually goes up. The trade-off is practical. A broader supplier pool can help speed and cost in early development, but it increases the OEM's burden to define requirements, verify output, and maintain traceability.
Teams that work in other regulated sectors often recognize the pattern from supplier management systems such as IATF 16949 certification requirements in automotive manufacturing. Medical device work applies that same discipline with more attention to risk, traceability, and documented design controls.
Why teams care before certification
The value shows up in everyday decisions, not just during an audit.
When the system is working, the project team can answer questions that directly affect schedule and technical credibility:
- Which drawing and BOM revision were used for this build
- What material, process, and finish were supplied
- Which inspection results apply to the parts used in testing
- Whether the supplier was qualified for this scope of work
- What changed between prototype, verification build, and released design
Those answers determine whether test data can be used with confidence or has to be repeated.
A common failure mode in medical device programs is not a single dimensional miss. It is an inability to prove which version was tested, how it was produced, what requirements applied at the time, and whether the build can be repeated under control. That gap creates rework, delayed verification, supplier disputes, and weak design transfer packages.
ISO 13485 matters because it ties the paper system to the physical build. It gives engineering, quality, and sourcing one operating model for deciding when a printed part is good enough, when a machined component needs tighter documentation, and when a low-volume run has to be treated like the start of production rather than "just prototyping."
Understanding the Key Requirements of ISO 13485
The full standard is broad, but a project engineering team usually feels four requirements most directly: risk management, documentation, process validation, and traceability. These aren't abstract quality topics. They shape what you buy, what you inspect, and what evidence you keep.

Risk control starts with design decisions
Risk management under ISO 13485 isn't something you bolt on after design freeze. It has to influence material selection, geometry, manufacturability, and inspection planning while the part is still changing.
A few common examples make this concrete:
- Tight internal corners in a machined plastic part may create tool access limits and dimensional variability.
- A cosmetic surface callout may be functional if the part interfaces with tissue, fluid, or sealing features.
- A printed prototype may be useful for fit checks but inappropriate for validation if the production material behavior is critical.
When teams miss this, they often generate data on parts that don't represent the eventual device well enough to support later decisions.
The Medical Device File has to match the physical part
ISO 13485:2016 requires each medical device family to maintain a Medical Device File with explicit specifications for manufacturing, inspection, packaging, and storage, and undefined preservation specs or missing inspection protocols directly correlate to non-conformance, as described in this Medical Device File explanation.
That requirement gets very practical for sourced components. If a machined part needs a defined surface finish, dimensional acceptance method, packaging method, or environmental handling condition, those details can't live only in someone's inbox or in a supplier's tribal knowledge.
For physical parts, the file often needs to align with records such as:
- Released drawings with critical dimensions and tolerances
- Inspection plans that state what is measured and how
- Surface finish requirements such as Ra or Rz targets when function depends on them
- Packaging and storage instructions that preserve device integrity
- Distribution controls if temperature, humidity, or handling conditions matter
If those controls aren't defined, you don't just have a documentation gap. You have a build consistency problem.
Process validation and traceability show up earlier than most teams expect
Some teams assume validation starts only at production scale. In practice, process validation thinking starts when you identify which manufacturing steps can't be fully assured by final inspection alone.
That's common in medical work. Surface treatment, cleaning, additive builds, bonding, molding, and complex machining setups can all introduce variables that aren't obvious by looking at the finished part.
Traceability matters just as much. You need to know what lot, revision, material condition, and process route produced the component used in a critical build.
A useful way to think about it is this short checklist:
| Requirement area | What engineering must define | What manufacturing must provide |
|---|---|---|
| Risk management | Critical features and failure-sensitive characteristics | Controls that reduce build variation |
| Documentation | Released specs, acceptance criteria, preservation needs | Records that match the released specs |
| Process validation | Which steps need controlled repeatability | Evidence that the process runs consistently |
| Traceability | What must be linked to the device history | Batch, lot, inspection, and revision records |
The standard becomes much easier to manage when the team stops viewing it as a clause list and starts viewing it as a chain from design intent to physical evidence.
ISO 13485 vs ISO 9001 and Regulatory Frameworks
A project team releases a prototype housing to a machine shop. The quote looks good, the tolerances look achievable, and the supplier has ISO 9001 certification. Then the questions start. Can they control revision changes after the first build? Can they keep lot traceability on the resin insert and passivation step? Can they show which inspection method was used on the parts that went into the verification build?
That is the point where the difference between ISO 9001, ISO 13485, and regulatory requirements stops being theoretical.
Why ISO 9001 does not answer the medical device question
ISO 9001 is a solid baseline for general quality management. It shows that a supplier has a structured system for handling processes, records, corrective action, and customer requirements. For CNC machining, molding, and finishing suppliers, that matters.
Medical device work usually needs more than that. ISO 13485 puts tighter expectations around document control, traceability, validation, complaint handling, risk-based controls, and the way outsourced processes are managed. Those differences show up fast in prototype and low-volume builds, where engineering changes are frequent and the temptation is to treat every run as informal.
A supplier with ISO 9001 may still be the right fit for an early concept model or a non-clinical fixture. That same supplier may be a poor fit for a design verification build if the part history, inspection evidence, and change control need to feed directly into your device records.
The same pattern exists in other industries. A supplier can have a mature sector-specific quality system and still not meet medical device expectations. That is why teams compare standards instead of treating any certification as universally interchangeable, which is also clear in work done under IATF 16949 certification for automotive manufacturing quality systems.
How ISO 13485 relates to FDA and EU requirements
ISO 13485 is a standard. FDA and EU requirements are legal obligations.
That distinction matters because teams often ask the wrong question. They ask whether a supplier is "ISO 13485 certified" when the better question is whether the supplier can support the controls your device and market require. Certification can help. It does not transfer regulatory responsibility away from the legal manufacturer.
For FDA-regulated products, the agency does not issue ISO 13485 certificates, and certification does not replace FDA inspection or compliance obligations. Under the QMSR approach, alignment between FDA quality system expectations and ISO 13485 is closer than it used to be, but the manufacturer still has to show the system works in practice, as explained in this FDA QMSR and ISO 13485 overview.
In the EU, the same practical lesson applies. A certificate may support market access and supplier qualification, but it does not remove the need to meet MDR requirements, maintain technical documentation, and control post-market responsibilities.
What this means during prototyping and supplier selection
For engineering teams, the practical difference is simple. ISO 9001 tells you a supplier likely has a general quality structure. ISO 13485 gives better confidence that the supplier understands medical-device-style controls. Regulatory compliance still sits with the OEM or legal manufacturer, especially for design decisions, risk files, and release decisions.
That affects sourcing choices:
- A cosmetic appearance model may only need a capable supplier with basic document control.
- A verification build usually needs stronger revision control, inspection records, and material traceability.
- A pilot run for clinical or regulatory use may require validated processes, formal nonconformance handling, and clearer evidence for each lot built.
Teams get into trouble when they treat all prototype parts the same. A one-off 3D print for ergonomic review does not carry the same quality burden as a machined implant instrument component used in formal testing.
Here is the comparison that usually clears it up:
| Aspect | ISO 9001:2015 | ISO 13485:2016 | FDA QMSR / EU MDR |
|---|---|---|---|
| Primary purpose | General quality management across industries | Quality management for medical device organizations | Legal requirements for placing and maintaining devices on the market |
| Scope of controls | Broad and flexible | More specific to device quality, traceability, and controlled records | Jurisdiction-specific obligations enforced by regulators or notified bodies |
| Risk focus | General process and customer satisfaction focus | Risk-based controls tied to device quality and patient safety | Risk management expected as part of regulatory compliance |
| Documentation depth | General quality documentation | Tighter control of device-related records and changes | Evidence must support legal compliance and product claims |
| Supplier relevance | Good baseline for capability | Better fit for suppliers supporting medical device builds | OEM remains responsible for outsourced process control |
| What it means for prototyping | Useful for early, lower-risk builds | Better suited to verification, pilot, and controlled low-volume work | Determines what evidence the OEM must retain and review |
The practical takeaway is to qualify suppliers by build purpose, not by certificate alone. If the parts will feed design verification, clinical evaluation, process validation, or a submission record, ask how the supplier handles revision control, traceability, inspections, and special processes. That answer matters more than the logo on the certificate.
An Implementation Checklist for ISO 13485
Implementation goes smoother when you treat it like a product launch project instead of a documentation exercise. The companies that struggle usually overbuild procedures before they've mapped how work is performed. The ones that do better start with scope, process ownership, and evidence flow.

Phase one sets the pace
Start with leadership alignment and a realistic scope. Decide which products, sites, and processes the system covers. If your organization already uses structured inspection practices such as first article inspection, map those into the QMS instead of creating duplicate paperwork.
The first pass should answer a few direct questions:
- Which processes already exist and operate effectively
- Where records are being created but not controlled
- Which outsourced processes affect device quality
- Where the biggest patient-safety or compliance risks sit
A gap assessment is useful only if it compares current behavior to required behavior. It shouldn't be a generic template review.
Build the system around real work
Most implementation effort lands in process definition, documentation, and training. Keep it practical. Write procedures that reflect how engineering releases drawings, how purchasing approves suppliers, how incoming inspection records results, and how nonconformances are handled.
The strongest implementations usually have these characteristics:
- Process owners are named. Quality can govern, but engineering, operations, and supply chain must own their workflows.
- Forms and records are simple. If a record takes too long to complete, people will create side systems.
- Training is role-based. Machinists, buyers, and design engineers don't need the same depth on every clause.
- Risk drives priority. Focus first on processes that can affect patient safety, performance, or regulatory evidence.
If a procedure doesn't match how work is really performed, the audit finding is already built in. It just hasn't been written yet.
Audit before the registrar does
Before an external audit, run the system long enough to generate real records. Then audit those records. Don't stop at whether a form exists. Check whether the information on the form would help reconstruct what happened.
A solid pre-certification rhythm usually includes:
- Internal audits that test implementation, not just document presence
- Management review with actual quality and process issues on the agenda
- Corrective actions closed with evidence, not verbal assurance
- Supplier files that show approval, monitoring, and clear expectations
The implementation target isn't a polished manual. It's a working system that people actively use under schedule pressure.
Supplier and Prototyping Implications
The standard's impact becomes tangible for sourcing teams. The question isn't only whether a supplier can machine or print the part. The primary question is whether the supplier can produce the part under a level of control that fits the build's purpose.

ISO 13485 requires design controls throughout development, so CNC and 3D-printed prototypes for many devices must go through verification and validation before moving to production. The standard also distinguishes design verification from design validation, with validation focused on intended use and often requiring clinical or performance evaluation for higher-risk devices, as explained in this design verification and validation discussion.
That has a direct effect on prototype sourcing. If the prototype is only for visual review, you can accept more flexibility. If the build will support formal testing, the manufacturing route, material condition, inspection evidence, and change control suddenly matter a lot more.
What to ask a prototype supplier
A useful supplier conversation is more detailed than “Are you ISO certified?”
Ask things like:
- Can you hold and document critical dimensions on this geometry
- Will the shop provide inspection results tied to the drawing revision
- How are lots, batches, or build records identified
- What happens if a programmer, machine, resin, or setup changes mid-project
- Can the supplier support both early prototypes and low-volume controlled builds
If you're comparing shops, a supplier with broad rapid prototyping capabilities can reduce handoff risk between early models and later controlled iterations. That matters because every supplier change introduces a new validation and documentation burden.
When supplier certification matters most
Not every supplier touching a program needs the same level of quality maturity. But certification becomes much more valuable when the supplier's output feeds formal development evidence or low-volume preproduction work.
Here's the trade-off in practical terms:
| Sourcing situation | What matters most |
|---|---|
| Early concept mockup | Speed, basic dimensional suitability, communication |
| Functional prototype for engineering learning | Repeatability, material clarity, useful inspection feedback |
| Verification build | Controlled revision use, traceable inspection, stable process |
| Validation or pilot build | Stronger supplier controls, documented consistency, change discipline |
A common mistake is using an informal prototype supplier deep into the program because the early parts arrived quickly. That works until the team needs traceable evidence or matching replacement builds.
The cheapest prototype is often the one you can keep using as the program matures. The expensive one is the part that has to be rebuilt under tighter controls because the original supplier left no usable trail.
Supplier choice is part of risk management. For medical devices, that isn't theory. It directly affects whether your test data stays credible.
The Audit and Certification Process Explained
A certification audit usually exposes problems that were built in months earlier. The trigger is often simple: an engineer orders verification parts from one shop, purchasing switches to another for the next build, and nobody updates the approved supplier record, drawing distribution list, or inspection plan. The audit finding shows up late. The control failure started much earlier on the shop floor.
By the time a company invites in a registrar, the quality system should already be part of daily work. Procedures alone will not carry the audit. Auditors want to see that design, purchasing, manufacturing, and quality teams are using the same controls the same way, especially where prototype builds turn into formal verification or pilot production.
What the initial certification cycle looks like
The certification path is straightforward, but it tests whether the system is real.
- Registrar selection. Choose a certification body that fits your product scope, markets, and audit expectations.
- Stage 1 review. The auditor reviews the quality system documentation and checks whether the company is ready for a full assessment.
- Stage 2 audit. The auditor samples actual implementation across departments, records, training, supplier controls, and operational evidence.
- Surveillance audits. The certification body returns on a scheduled basis to confirm the system is still being followed and maintained.
- Recertification. The company goes through a broader review again at the end of the certification cycle.
The practical mistake is treating Stage 1 as a paperwork exercise and Stage 2 as the definitive test. In medical device work, both matter. If the documented process says prototype suppliers are handled informally, but the same suppliers are already making parts used in design verification, the inconsistency will draw attention fast.
What auditors usually test in practice
Auditors follow traceability through real jobs. They pick a design change, a purchase order, a nonconformance, or a released lot and ask the team to show how decisions were controlled.
For a medical device manufacturer, that usually includes:
- Design inputs, outputs, reviews, and change history
- Supplier qualification, approval, and ongoing monitoring
- Inspection records tied to acceptance decisions
- Control of nonconforming product and corrective action
- Training records matched to the work people perform
- Management review evidence that led to decisions or follow-up actions
Prototype and low-volume builds are often where weak controls show up first. A CNC supplier may have made acceptable concept parts with minimal documentation. That same supplier becomes a much bigger audit concern if the team later uses those parts in verification without defined revision control, material traceability, or acceptance criteria. The issue is not that prototype work is forbidden. The issue is whether the company defined what level of control was needed for the intended use.
I have seen auditors spend more time on one build history than on a stack of procedures. If a team can show the released drawing revision, supplier approval status, incoming inspection result, deviation record, and disposition decision without hunting through email, the audit usually moves well. If that evidence is scattered across shared drives, inboxes, and tribal knowledge, the discussion gets expensive.
The strongest audit preparation is operational discipline. Run internal audits against real records. Review whether prototype suppliers are being used beyond their original purpose. Check that engineers, buyers, and inspectors are all working from the same revision and the same supplier status. That is what makes certification maintainable after the certificate arrives.
If your team needs a manufacturing partner that understands how prototype parts, inspection records, and low-volume production fit inside a regulated development workflow, LC Proto supports CNC machining, 3D printing, and short-run manufacturing with ISO-based quality systems suited to medical device programs.


